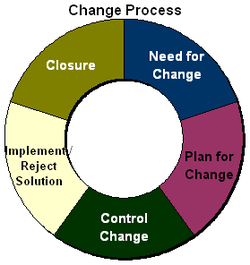
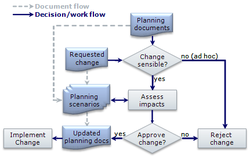
In my last post, I described change control process in general and I mentioned that in the regulated industries, manufactures are required to use a change control procedure. I am going to describe this change control procedure in this post.
A change control procedure is usually one of standard operating procedures (SOP's). It usually includes a change control form. Some companies also use change request forms for suggested changes. This procedure usually includes the following components:
Identification
The identification of the changed device, assembly, component, labeling, packaging, software, process, procedure, manufacturing material, and any other related item or document. The change control form has blank spaces for recording this data.
Effective Date
The effective date of the change which is usually a completion date, or an action to be performed when a specific event occurs, such as "implement the change when the new part is installed, validated, and operational." The blank on the change control form for recording the effective date should not be left empty.
Responsibility
The change procedure should state which department or designee is responsible for each function to be performed.
Revision Number
The change procedure should describe the way the revision level is to be incremented. It is common practice to use sequential numbers for revisions.
Communication
The change procedure should describe the communication of changes to all affected parties such as production, purchasing, contractors, suppliers, etc. As appropriate, the document might include activities that apply to internal operations. Examples are employee training, rework, or disposition of in-process assemblies, use of revised drawings and/or procedures, and disposition of old documents.
Updating Documentation
The change procedure should cover updating of primary and secondary documentation such as instruction manuals. Usually there are no problems with updating or revising primary documentation -- in fact, that is a major reason the given change order is being processed. In contrast, it is rather easy to forget that related secondary documents such as component drawings, instruction manuals or packaging require revision if affected by a given change. The use of a good change control form can alleviate this problem.
Documentation Distribution
Revised documentation should be distributed to persons responsible for the operations affected by the change and old documents removed and filed or discarded, as appropriate. After updated documents have been approved, these documents have to be made available at all locations for which they are designated, used, or otherwise necessary, and all obsolete documents have to be promptly removed from all points of use or otherwise prevented from unintended use.
Remedial Actions
Certain changes may require remedial action. Changes of this nature should be addressed in the change control procedure.
Regulatory Submissions
There may be changes may that require a regulatory submission. The change control procedure should specify if regulatory submissions should be considered when making a change.
Business Factors
The change procedure should also cover other factors such as financial impact, modification of sales literature, update of products in commercial distribution, etc.
Quality Assurance Review
The change procedure should cover if the quality assurance review is required for the change.
This change control procedure is also used for document control.
Changes to documents have to be reviewed and approved by an individual(s) in the same function or organization that performed the original review and approval of these documents unless there is a specific designation that states otherwise. These approved changes have to be communicated to the appropriate personnel in a timely manner. A company has to maintain records of changes to documents.
Change control for documents should include:
A change control procedure is usually one of standard operating procedures (SOP's). It usually includes a change control form. Some companies also use change request forms for suggested changes. This procedure usually includes the following components:
Identification
The identification of the changed device, assembly, component, labeling, packaging, software, process, procedure, manufacturing material, and any other related item or document. The change control form has blank spaces for recording this data.
Effective Date
The effective date of the change which is usually a completion date, or an action to be performed when a specific event occurs, such as "implement the change when the new part is installed, validated, and operational." The blank on the change control form for recording the effective date should not be left empty.
Responsibility
The change procedure should state which department or designee is responsible for each function to be performed.
Revision Number
The change procedure should describe the way the revision level is to be incremented. It is common practice to use sequential numbers for revisions.
Communication
The change procedure should describe the communication of changes to all affected parties such as production, purchasing, contractors, suppliers, etc. As appropriate, the document might include activities that apply to internal operations. Examples are employee training, rework, or disposition of in-process assemblies, use of revised drawings and/or procedures, and disposition of old documents.
Updating Documentation
The change procedure should cover updating of primary and secondary documentation such as instruction manuals. Usually there are no problems with updating or revising primary documentation -- in fact, that is a major reason the given change order is being processed. In contrast, it is rather easy to forget that related secondary documents such as component drawings, instruction manuals or packaging require revision if affected by a given change. The use of a good change control form can alleviate this problem.
Documentation Distribution
Revised documentation should be distributed to persons responsible for the operations affected by the change and old documents removed and filed or discarded, as appropriate. After updated documents have been approved, these documents have to be made available at all locations for which they are designated, used, or otherwise necessary, and all obsolete documents have to be promptly removed from all points of use or otherwise prevented from unintended use.
Remedial Actions
Certain changes may require remedial action. Changes of this nature should be addressed in the change control procedure.
Regulatory Submissions
There may be changes may that require a regulatory submission. The change control procedure should specify if regulatory submissions should be considered when making a change.
Business Factors
The change procedure should also cover other factors such as financial impact, modification of sales literature, update of products in commercial distribution, etc.
Quality Assurance Review
The change procedure should cover if the quality assurance review is required for the change.
This change control procedure is also used for document control.
Changes to documents have to be reviewed and approved by an individual(s) in the same function or organization that performed the original review and approval of these documents unless there is a specific designation that states otherwise. These approved changes have to be communicated to the appropriate personnel in a timely manner. A company has to maintain records of changes to documents.
Change control for documents should include:
- identification of the affected documents;
- a description of the change;
- revision number
- the signature of the approving individual(s);
- the approval date;
- the date when the change becomes effective.


 RSS Feed
RSS Feed
